Mitral Valve Prolapse
|
Clinical Problem: Mitral valve prolapse (MVP) is a common valvular lesion that affects 2-5% of the general population. In this lesion, the mitral valve leaflet and chordal tissue has inherent changes in its biological make-up. These changes impact leaflet closure, by inducing billowing of one or both of the leaflets into the left atrium, or rupture of one or more chordae that causes localized loss of leaflet coaptation. Increasing evidence is pointing to specific genetic mutations that perturb the mitral valve biology in mice and zebrafish. However, since these biological changes manifest in the 4th to 8th decade of life in humans, it is unclear if genetic mutations are the only cause of this lesion. Our laboratory is interested in investigating the interaction between genetic mutations and mitral valve bio-mechanics in the development of MVP.
|
|
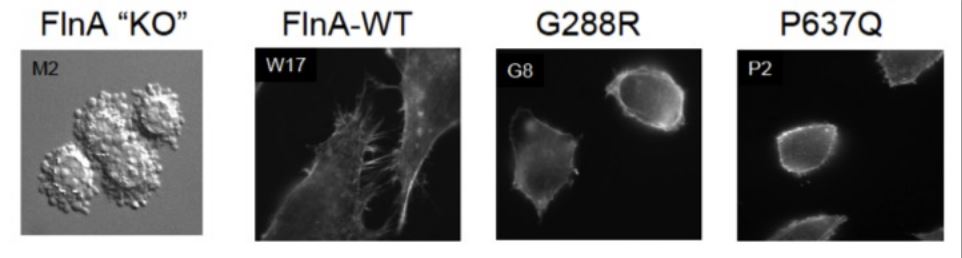
Genetics of MVP: An X-linked form of non-syndromic MVP was recently reported, identifying mis-sense mutations in a gene in an 8-cM interval on chromosome Xq28, which encodes Filamin-A (FLNA). In one large family of 91 living members, a missense mutation with a C to A transition at nucleotide 1910 in exon 13 was reported, which demonstrated a substitution of a proline (P) for a glutamine (Q) at amino acid 637 (P637Q). In another family, a G to A transition at nucleotide 862 in exon 5 was reported, resulting in a substitution of glycine (G) for arginine (R) at amino acid 288. In a third family, a T to A transition at nucleotide 2132 in exon 14 was identified, which transcribes to replacement of valine (V) for asparatic acid (D) at amino acid 711 (V711D). In a final family, a 1944bp genomic deletion in exons 16 to 19 was discovered, inducing a 546bp deletion of 182 amino acids in the protein (V761 to Q943). FLNA is a large dimeric actin-binding cytoskeletal protein, where it governs formation of dynamic orthogonal actin filament networks in response to extracellular cues, regulates signaling pathways that enable cellular sensing of extracellular mechanical forces and substrate stiffness, and forms the dynamic machinery required for cellular motility. Our laboratory is interested in investigating if MVP relevant FLNA mutations alter cellular architecture, morphology and expression of transmembrane proteins that are essential for mechano-transduction, and alter the mitral valve micro mechanics.
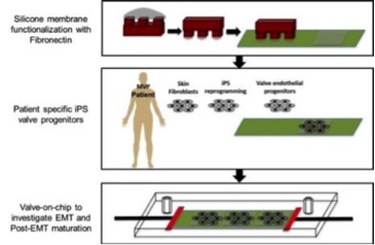
Interaction between genetics and mechanics in MVP pathogenesis: MVP is an interesting lesion, which has beginnings in genetic mutations, but its complete phenotype does not manifest until several decades after birth. Our lab is interested in understanding the process of "biological fatigue"- i.e. mutations leading to progressive biological weakening in the valve leaflets. We are developing a 'miniature valve model' in which controlled mechanical strain and fluid shear can be imposed on mitral valve cells with mutations. Our cells are sourced from patients with the mutations, where their skin fibroblasts are reprogrammed into iPS cells and induced to differentiate into valve endothelial cells and then endothelial-to-mesenchymal transition to form valve interstitial cells. Our hope is that this approach would help us understand the interaction between mechanics and genetics at each level of valvulogenesis and valve maturation.